



Biochip en lugar de análises xenéticas

As asociacións de hipercolesterolemia familiar levan anos loitando contra esta secuela. Os obxectivos marcados até o momento foron dous: abaratar o tratamento e facilitar a detección da enfermidade. Desta forma, nuns anos preténdese diagnosticar e o máis axiña posible a todas as persoas que poden padecer a enfermidade, xa que moitas veces, tras sufrir un infarto, sábese que o paciente tiña una hipercolesterolemia familiar.
Parece que ambos están en vías de alcanzar. Por unha banda, desde que comezou o ano os medicamentos son máis baratos e en canto ao diagnóstico avanzouse moito, xa que se está comercializando un biochip de análise xenética. A empresa vasca Medplant Genetics/Progenika desenvolveu un biochip denominado Lipochip®. Este biochip permite un diagnóstico rápido e seguro.
Posibilidade de diagnóstico a tempo
Até o momento utilizouse una táboa de criterios da Organización Mundial da Saúde paira detectar a hipercolesterolemia familiar (FH), na que se teñen en conta os niveis de colesterol en sangue, casos de infarto na familia, etc., pero este método non é do todo fiable. O método máis preciso, sen dúbida, é o de realizar unha análise xenética, pero é longo. A partir de agora, con todo, o biochip realizado especificamente paira a detección de FH facilitará enormemente o traballo: o resultado pódese recoller ao día seguinte da extracción dunha mostra de sangue ao paciente.
Debido ás mutacións dun xene FH, o biochip foi deseñado paira detectar estas mutacións. Consiste basicamente nunha placa de vidro coas secuencias auxiliares correspondentes a cada una das mutacións causantes da FH, os oligonucleótidos. Paira a análise extráese o ADN da mostra de sangue do paciente e colócase na placa marcado por fluorescencia. A luz láser permite ver en que punto únese a mostra con maior forza á placa e, en consecuencia, determinar a mutación do paciente si tivésea.
O diagnóstico é seguro, é dicir, si detéctase una mutación pódese dicir sen dúbida que o paciente ten FH. Con todo, a non detección de mutacións non significa que o paciente non teña FH senón que non ten mutacións que o biochip detecta.
De feito, a enfermidade é causada por mutacións nun xene determinado que se atopa no brazo curto do cromosoma 19. E en todo o mundo coñécense unhas 700 mutacións que provocan FH; por dicilo dalgunha maneira, as mutacións están xeograficamente separadas, é dicir, en cada pobo aparecen determinadas mutacións.
Nos territorios nos que se moveron moitos pobos, o patrimonio xenético destes pobos quedou no xenoma dos seus habitantes, e as mutacións forman parte dese patrimonio. Por exemplo, na poboación española coñécense 181 mutacións, das cales 70 non se atoparon en ningún outro lugar. Lipochip® está especialmente deseñado paira detectar mutacións neste territorio. Detecta 154 delas e en breve engadiranse os oligonucleótidos correspondentes a outras 25 mutacións. Noutros territorios poden aparecer mutacións iguais ou moi diferentes, polo que para que o biochip sexa útil será necesario adaptarse ás mutacións locais.
Outra gran vantaxe deste chip é que se sabe cal é a mutación exacta do paciente, o que permite que o médico poida prescribir o tratamento máis adecuado. De feito, non todas as mutacións teñen a mesma incidencia no metabolismo do paciente, algunhas son máis graves que outras e requiren un tratamento máis eficaz.
Colesterol, efectos e axentes
A FH é una enfermidade do metabolismo do colesterol que pasa dunha xeración a outra a través da herdanza autosómica dominante. Paira coñecer a influencia destas mutacións herdadas hai que ir ao fígado. De feito, o xene de FH codifica o receptor de LDL, que é o receptor que as células hepáticas teñen paira regular o nivel de colesterol en sangue. Grazas a eles, si o colesterol en sangue é excesivo, sácase do sangue e se interna na célula, diminuíndo así os niveis de colesterol no sangue.
Pola contra, si os receptores LDL non funcionan correctamente, o colesterol en sangue é excesivo e aos poucos vaise acumulando nas paredes dos vasos sanguíneos e producindo arteriosclerosis. Esta acumulación dificulta a circulación do sangue e, entre outras cousas, pode formar coácidos, e si non chega suficiente osíxeno ás células prodúcese un infarto. Por iso, non é de estrañar que uno dos principais causantes de enfermidades do corazón sexa o colesterol.
Con todo, non todos os casos de FH son iguais e os problemas metabólicos poden ser moi diferentes. No caso máis grave, o enfermo recibiu o xene da FH de ambos os proxenitores, é homocigoto. Nestes casos, o fígado non recibe colesterol, polo que o enfermo ten exceso de colesterol no seu sangue desde o nacemento. Ademais, atópase a concentracións moi elevadas, superiores a 1000 mg/dl.
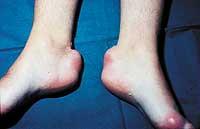
O homocigoto FH é o máis difícil de tratar e en moitos casos é necesario utilizar medicamentos de forma combinada paira minimizar os efectos secundarios. Neste caso, o colesterol tamén se acumula debaixo da pel (formando estruturas denominadas xantalemas) e nos ligamentos (formando xantemas).
O habitual, con todo, é una mutación heterocigota, na que a mutación é o único alelo do xene. O fígado non regula ben os niveis de colesterol, xa que non ten suficientes receptores nas células. Tratamento máis sinxelo que a mutación homocigota, xa que a concentración de colesterol tamén é menor, entre 300 e 500 mg/dl. Con todo, a concentración sanguínea de colesterol atópase por encima dos 120-240 mg/dl habituais, sendo imprescindible o tratamento.
Desgraciadamente, a FH non pode curarse, pero polo menos cun tratamento adecuado o paciente pode levar una vida normal e longa. Grazas a este lipochip® espérase que nuns anos todos os pacientes sexan diagnosticados. Será un proxecto caro, pero rendible paira as organizacións sanitarias co tempo, xa que os ingresos hospitalarios paira tratar os ataques cardíacos, ictus, murmurios e demais enfermidades que non favorezan o seu diagnóstico e tratamento serían moito máis custosos.
Hipercolesterolemia familiar en datosA alta taxa de colesterol non é rara na actualidade. Esta hipercolesterolemia atribúese a unha dieta rica en graxas e á non realización de exercicio físico. Pero nalgúns casos, a pesar de una dieta adecuada e de practicar deporte, a taxa de colesterol en sangue é elevada. Isto é debido á hipercolesterolemia familiar e é conveniente realizar un diagnóstico seguro canto antes. En Euskal Herria hai unhas seis mil persoas que padecen esta enfermidade xenética, pero a maioría non o saben. Só un 30% dos pacientes están detectados. O resto leva una vida normal, sen un tratamento adecuado paira reducir a taxa de colesterol, co consecuente risco de sufrir un infarto. Lamentablemente, a maioría dos casos de FH diagnostícanse despois de una emerxencia cardíaca, que en moitos casos é demasiado tarde, xa que o paciente faleceu. As estatísticas sobre pacientes con FH son realmente dramáticas: a esperanza de vida é entre 20 e 30 anos menor que o resto. Ademais, os homes teñen máis risco de sufrir un infarto que as mulleres: O 75% dos homes con FH sofre un infarto antes dos 60 anos. |
Buletina
Bidali zure helbide elektronikoa eta jaso asteroko buletina zure sarrera-ontzian







