



Biochip en lugar de análisis genéticos

Las asociaciones de hipercolesterolemia familiar llevan años luchando contra esta lacra. Los objetivos marcados hasta el momento han sido dos: abaratar el tratamiento y facilitar la detección de la enfermedad. De esta forma, en unos años se pretende diagnosticar y a la mayor brevedad posible a todas las personas que pueden padecer la enfermedad, ya que muchas veces, tras sufrir un infarto, se sabe que el paciente tenía una hipercolesterolemia familiar.
Parece que ambos están en vías de alcanzar. Por un lado, desde que comenzó el año los medicamentos son más baratos y en cuanto al diagnóstico se ha avanzado mucho, ya que se está comercializando un biochip de análisis genético. La empresa vasca Medplant Genetics/Progenika ha desarrollado un biochip denominado Lipochip®. Este biochip permite un diagnóstico rápido y seguro.
Posibilidad de diagnóstico a tiempo
Hasta el momento se ha utilizado una tabla de criterios de la Organización Mundial de la Salud para detectar la hipercolesterolemia familiar (FH), en la que se tienen en cuenta los niveles de colesterol en sangre, casos de infarto en la familia, etc., pero este método no es del todo fiable. El método más preciso, sin duda, es el de realizar un análisis genético, pero es largo. A partir de ahora, sin embargo, el biochip realizado específicamente para la detección de FH facilitará enormemente el trabajo: el resultado se puede recoger al día siguiente de la extracción de una muestra de sangre al paciente.
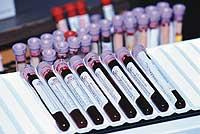
Debido a las mutaciones de un gen FH, el biochip ha sido diseñado para detectar estas mutaciones. Consiste básicamente en una placa de vidrio con las secuencias auxiliares correspondientes a cada una de las mutaciones causantes de la FH, los oligonucleótidos. Para el análisis se extrae el ADN de la muestra de sangre del paciente y se coloca en la placa marcado por fluorescencia. La luz láser permite ver en qué punto se une la muestra con mayor fuerza a la placa y, en consecuencia, determinar la mutación del paciente si la tuviera.
El diagnóstico es seguro, es decir, si se detecta una mutación se puede decir sin duda que el paciente tiene FH. Sin embargo, la no detección de mutaciones no significa que el paciente no tenga FH sino que no tiene mutaciones que el biochip detecta.
De hecho, la enfermedad es causada por mutaciones en un gen determinado que se encuentra en el brazo corto del cromosoma 19. Y en todo el mundo se conocen unas 700 mutaciones que provocan FH; por decirlo de alguna manera, las mutaciones están geográficamente separadas, es decir, en cada pueblo aparecen determinadas mutaciones.
En los territorios en los que se han movido muchos pueblos, el patrimonio genético de estos pueblos ha quedado en el genoma de sus habitantes, y las mutaciones forman parte de ese patrimonio. Por ejemplo, en la población española se conocen 181 mutaciones, de las cuales 70 no se han encontrado en ningún otro lugar. Lipochip® está especialmente diseñado para detectar mutaciones en este territorio. Detecta 154 de ellas y en breve se añadirán los oligonucleótidos correspondientes a otras 25 mutaciones. En otros territorios pueden aparecer mutaciones iguales o muy diferentes, por lo que para que el biochip sea útil será necesario adaptarse a las mutaciones locales.
Otra gran ventaja de este chip es que se sabe cuál es la mutación exacta del paciente, lo que permite que el médico pueda prescribir el tratamiento más adecuado. De hecho, no todas las mutaciones tienen la misma incidencia en el metabolismo del paciente, algunas son más graves que otras y requieren un tratamiento más eficaz.
Colesterol, efectos y agentes
La FH es una enfermedad del metabolismo del colesterol que pasa de una generación a otra a través de la herencia autosómica dominante. Para conocer la influencia de estas mutaciones heredadas hay que ir al hígado. De hecho, el gen de FH codifica el receptor de LDL, que es el receptor que las células hepáticas tienen para regular el nivel de colesterol en sangre. Gracias a ellos, si el colesterol en sangre es excesivo, se saca de la sangre y se interna en la célula, disminuyendo así los niveles de colesterol en la sangre.
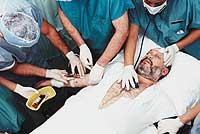
Por el contrario, si los receptores LDL no funcionan correctamente, el colesterol en sangre es excesivo y poco a poco se va acumulando en las paredes de los vasos sanguíneos y produciendo arteriosclerosis. Esta acumulación dificulta la circulación de la sangre y, entre otras cosas, puede formar coácidos, y si no llega suficiente oxígeno a las células se produce un infarto. Por ello, no es de extrañar que uno de los principales causantes de enfermedades del corazón sea el colesterol.
Sin embargo, no todos los casos de FH son iguales y los problemas metabólicos pueden ser muy diferentes. En el caso más grave, el enfermo ha recibido el gen de la FH de ambos progenitores, es homocigoto. En estos casos, el hígado no recibe colesterol, por lo que el enfermo tiene exceso de colesterol en su sangre desde el nacimiento. Además, se encuentra a concentraciones muy elevadas, superiores a 1000 mg/dl.
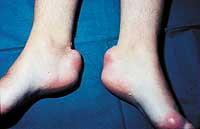
El homocigoto FH es el más difícil de tratar y en muchos casos es necesario utilizar medicamentos de forma combinada para minimizar los efectos secundarios. En este caso, el colesterol también se acumula debajo de la piel (formando estructuras denominadas xantalemas) y en los ligamentos (formando xantemas).
Lo habitual, sin embargo, es una mutación heterocigota, en la que la mutación es el único alelo del gen. El hígado no regula bien los niveles de colesterol, ya que no tiene suficientes receptores en las células. Tratamiento más sencillo que la mutación homocigota, ya que la concentración de colesterol también es menor, entre 300 y 500 mg/dl. Sin embargo, la concentración sanguínea de colesterol se encuentra por encima de los 120-240 mg/dl habituales, siendo imprescindible el tratamiento.
Desgraciadamente, la FH no puede curarse, pero al menos con un tratamiento adecuado el paciente puede llevar una vida normal y larga. Gracias a este lipochip® se espera que en unos años todos los pacientes hayan sido diagnosticados. Será un proyecto caro, pero rentable para las organizaciones sanitarias con el tiempo, ya que los ingresos hospitalarios para tratar los ataques cardíacos, ictus, murmullos y demás enfermedades que no favorezcan su diagnóstico y tratamiento serían mucho más costosos.
Hipercolesterolemia familiar en datosLa alta tasa de colesterol no es rara en la actualidad. Esta hipercolesterolemia se atribuye a una dieta rica en grasas y a la no realización de ejercicio físico. Pero en algunos casos, a pesar de una dieta adecuada y de practicar deporte, la tasa de colesterol en sangre es elevada. Esto es debido a la hipercolesterolemia familiar y es conveniente realizar un diagnóstico seguro lo antes posible. En Euskal Herria hay unas seis mil personas que padecen esta enfermedad genética, pero la mayoría no lo saben. Sólo un 30% de los pacientes están detectados. El resto lleva una vida normal, sin un tratamiento adecuado para reducir la tasa de colesterol, con el consiguiente riesgo de sufrir un infarto. Lamentablemente, la mayoría de los casos de FH se diagnostican después de una emergencia cardiaca, que en muchos casos es demasiado tarde, ya que el paciente ha fallecido. Las estadísticas sobre pacientes con FH son realmente dramáticas: la esperanza de vida es entre 20 y 30 años menor que el resto. Además, los hombres tienen más riesgo de sufrir un infarto que las mujeres: El 75% de los hombres con FH sufre un infarto antes de los 60 años. |
Buletina
Bidali zure helbide elektronikoa eta jaso asteroko buletina zure sarrera-ontzian







