



Biochip au lieu de l'analyse génétique

Les associations d'hypercholestérolémie familiale luttent depuis des années contre ce fléau. Les objectifs fixés jusqu'à présent ont été deux: couvrir le traitement et faciliter la détection de la maladie. Ainsi, dans quelques années on prétend diagnostiquer et le plus rapidement possible toutes les personnes qui peuvent souffrir de la maladie, car souvent, après avoir subi un infarctus, on sait que le patient avait une hypercholestérolémie familiale.
Il semble que les deux sont en voie d'atteindre. D'une part, depuis le début de l'année les médicaments sont moins chers et en ce qui concerne le diagnostic a beaucoup progressé, car un biochip d'analyse génétique est commercialisé. La société basque Medplant Genetics/Progenika a développé un biochip appelé Lipochip®. Ce biochip permet un diagnostic rapide et sûr.
Possibilité de diagnostic à temps
Jusqu'à présent, une table de critères de l'Organisation mondiale de la santé a été utilisée pour détecter l'hypercholestérolémie familiale (FH), dans laquelle on tient compte des niveaux de cholestérol sanguin, des cas d'infarctus dans la famille, etc., mais cette méthode n'est pas tout à fait fiable. La méthode la plus précise, sans doute, est de réaliser une analyse génétique, mais elle est longue. A partir de maintenant, cependant, le biochip réalisé spécifiquement pour la détection de FH facilitera grandement le travail: le résultat peut être recueilli le lendemain de l'extraction d'un échantillon de sang au patient.
En raison des mutations d'un gène FH, le biochip a été conçu pour détecter ces mutations. Il se compose essentiellement d'une plaque de verre avec les séquences auxiliaires correspondant à chacune des mutations causantes de la FH, les oligonucléotides. Pour l'analyse, l'ADN est extrait de l'échantillon de sang du patient et placé sur la plaque marquée par fluorescence. La lumière laser permet de voir à quel point l'échantillon est relié avec une plus grande force à la plaque et, par conséquent, de déterminer la mutation du patient s'il l'avait.
Le diagnostic est sûr, c'est à dire, si une mutation est détectée, vous pouvez certainement dire que le patient a FH. Cependant, la non-détection de mutations ne signifie pas que le patient n'a pas de FH mais n'a pas de mutations que le biochip détecte.
En fait, la maladie est causée par des mutations dans un gène particulier qui se trouve dans le bras court du chromosome 19. Et dans le monde entier, on connaît environ 700 mutations qui provoquent le FH; pour ainsi dire, les mutations sont géographiquement séparées, c'est-à-dire que dans chaque village apparaissent certaines mutations.
Dans les territoires où beaucoup de peuples se sont déplacés, le patrimoine génétique de ces peuples est resté dans le génome de leurs habitants, et les mutations font partie de ce patrimoine. Par exemple, la population espagnole connaît 181 mutations, dont 70 n'ont pas été trouvées ailleurs. Lipochip® est spécialement conçu pour détecter les mutations dans ce territoire. Il en détecte 154 et ajoutera bientôt les oligonucléotides correspondant à 25 autres mutations. Dans d'autres territoires peuvent apparaître des mutations égales ou très différentes, donc pour que le biochip soit utile il faudra s'adapter aux mutations locales.
Un autre grand avantage de cette puce est qu'il est connu quelle est la mutation exacte du patient, permettant au médecin de prescrire le traitement le plus approprié. En fait, toutes les mutations n'ont pas la même incidence sur le métabolisme du patient, certaines sont plus graves que d'autres et nécessitent un traitement plus efficace.
Cholestérol, effets et agents
La FH est une maladie de métabolisme de cholestérol qui passe d'une génération à l'autre par l'héritage autosomique principal. Pour connaître l'influence de ces mutations héritées, il faut aller au foie. En fait, le gène de FH codifie le récepteur de LDL, qui est le récepteur que les cellules du foie ont pour régler le niveau de cholestérol sanguin. Grâce à eux, si le cholestérol sanguin est excessif, il est retiré du sang et se trouve dans la cellule, diminuant ainsi le taux de cholestérol dans le sang.
En revanche, si les récepteurs LDL ne fonctionnent pas correctement, le cholestérol sanguin est excessif et s'accumule progressivement dans les parois des vaisseaux sanguins et produit une artériosclérose. Cette accumulation entrave la circulation du sang et, entre autres, peut former des coacides, et si l'oxygène ne suffit pas aux cellules, un infarctus se produit. Il n'est donc pas surprenant que l'un des principaux facteurs de maladies cardiaques soit le cholestérol.
Cependant, tous les cas de FH ne sont pas égaux et les problèmes métaboliques peuvent être très différents. Dans le cas le plus grave, le malade a reçu le gène de la FH des deux parents, est homozygote. Dans ces cas, le foie ne reçoit pas de cholestérol, de sorte que le malade a excès de cholestérol dans son sang depuis la naissance. En outre, il se trouve à des concentrations très élevées, supérieures à 1000 mg/dl.
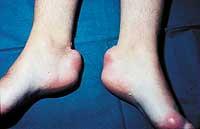
L'homozygote FH est le plus difficile à traiter et il est souvent nécessaire d'utiliser des médicaments combinés pour minimiser les effets secondaires. Dans ce cas, le cholestérol s'accumule également sous la peau (formant des structures appelées xanthalèmes) et dans les ligaments (formant des xanthèmes).
L'habitude, cependant, est une mutation hétérozygote, dans laquelle la mutation est le seul allèle du gène. Le foie ne régule pas bien les niveaux de cholestérol, car il n'a pas assez de récepteurs dans les cellules. Traitement plus simple que la mutation homozygote, car la concentration de cholestérol est également inférieure, entre 300 et 500 mg/dl. Cependant, la concentration sanguine de cholestérol est supérieure à 120-240 mg/dl habituels, le traitement étant indispensable.
Malheureusement, la FH ne peut pas être guérie, mais au moins avec un traitement approprié le patient peut mener une vie normale et longue. Grâce à ce lipochip®, tous les patients ont été diagnostiqués dans quelques années. Ce sera un projet coûteux, mais rentable pour les organismes de santé au fil du temps, car les revenus hospitaliers pour traiter les crises cardiaques, ictus, murmures et autres maladies qui ne favorisent pas leur diagnostic et leur traitement seraient beaucoup plus coûteux.
Hypercholestérolémie familiale en donnéesLe taux élevé de cholestérol n'est pas rare aujourd'hui. Cette hypercholestérolémie est attribuée à un régime riche en graisses et à la non-exercice physique. Mais dans certains cas, malgré une alimentation adéquate et le sport, le taux de cholestérol sanguin est élevé. Ceci est dû à l'hypercholestérolémie familiale et il est commode de réaliser un diagnostic sûr dès que possible. En Euskal Herria, il y a environ six mille personnes qui souffrent de cette maladie génétique, mais la plupart ne le savent pas. Seulement 30% des patients sont détectés. Le reste mène une vie normale, sans traitement adéquat pour réduire le taux de cholestérol, ce qui entraîne un risque d'infarctus. Malheureusement, la plupart des cas de FH sont diagnostiqués après une urgence cardiaque, qui est souvent trop tard, puisque le patient est décédé. Les statistiques sur les patients atteints de FH sont vraiment dramatiques: l'espérance de vie est entre 20 et 30 ans inférieure au reste. En outre, les hommes ont plus de risque de subir un infarctus que les femmes: 75% des hommes atteints de FH souffrent d'un infarctus avant 60 ans. |
Buletina
Bidali zure helbide elektronikoa eta jaso asteroko buletina zure sarrera-ontzian







