



Reciclar proteïnes, clau per a estar sa
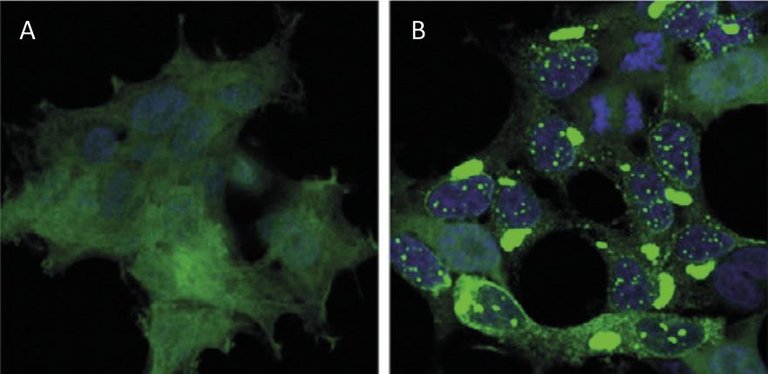
El reciclatge de proteïnes a nivell cel·lular té un impacte similar al del reciclatge de residus a nivell mundial. Les proteïnes són les principals molècules que garanteixen el funcionament de les cèl·lules. Són responsables de dur a terme reaccions químiques, regular els gens, transportar molècules i mantenir l'estructura cel·lular. Cada cèl·lula conté milers de proteïnes que a més es formen de forma molt dinàmica. De fet, per a respondre a les necessitats de la cèl·lula actual o als estímuls externs, s'està produint i destruint constantment proteïnes de diferents tipus cel·lulars. Per tant, és imprescindible mantenir l'equilibri entre aquests dos processos adversos per a garantir el correcte funcionament de la cèl·lula. Per això, no és d'estranyar que molts dels processos de síntesis i degradació de proteïnes que assoten la nostra societat estiguin en l'origen de moltes malalties. Entre les malalties desenvolupades com l'Alzheimer, el Parkinson i l'Huntington, totes elles molt conegudes [1,2] (Figura 1).
Proteïnes: ben plegades o acabades.
La informació per a la producció de proteïnes està emmagatzemada en els gens. En la cadena d'ADN s'especifica quan i quina proteïna es generarà. L'estructura d'una proteïna que s'extreu de la fàbrica de producció de proteïnes, és a dir, dels ribosomes, és equivalent a un collaret de perles amb aminoàcids. No obstant això, aquesta proteïna, per a ser funcional, ha de tenir una determinada estructura tridimensional. En la cèl·lula es poden trobar 20 tipus d'aminoàcids i la seqüència d'aminoàcids de cada proteïna determina l'estructura funcional tridimensional, és a dir, l'estructura nativa d'aquesta proteïna. Els requisits cel·lulars són molt especials, per la qual cosa les proteïnes necessiten sovint l'ajuda de proteïnes denominades chaperones moleculars per a obtenir una estructura nativa. Els éssers humans tenim més de cent chapperones, cadascun dels quals s'encarrega del procés de plegat de centenars de proteïnes ituales. Quan l'activitat dels txaperones està danyada per les mutacions, el procés de plegat de les proteïnes diana no és l'adequat, per la qual cosa aquestes proteïnes no són funcionals. Si no es degraden, s'acumulen en la cèl·lula causant greus danys [4] (Figura 2).
Per exemple, les mutacions ocorregudes en el txaperón denominat HSPB1 provoquen una esclerosi lateral amiotròfica i una neuropatía greu denominada Charcot Marie Tooth [5,6]. I els que tenen el chaperón XIP mutat desenvolupen la síndrome de Gordon Holmes, un mal únic que causa hipogonadismo i problemes neurològics.
Però en tots els casos en els quals les proteïnes es dobleguen malament i s'acumulen patològicament, la culpa no és dels txaperos. En alguns casos, a causa de les mutacions que es produeixen en el genoma, les proteïnes es produeixen amb més aminoàcids dels necessaris, la qual cosa impedeix obtenir l'estructura nativa corresponent (Figura 2).
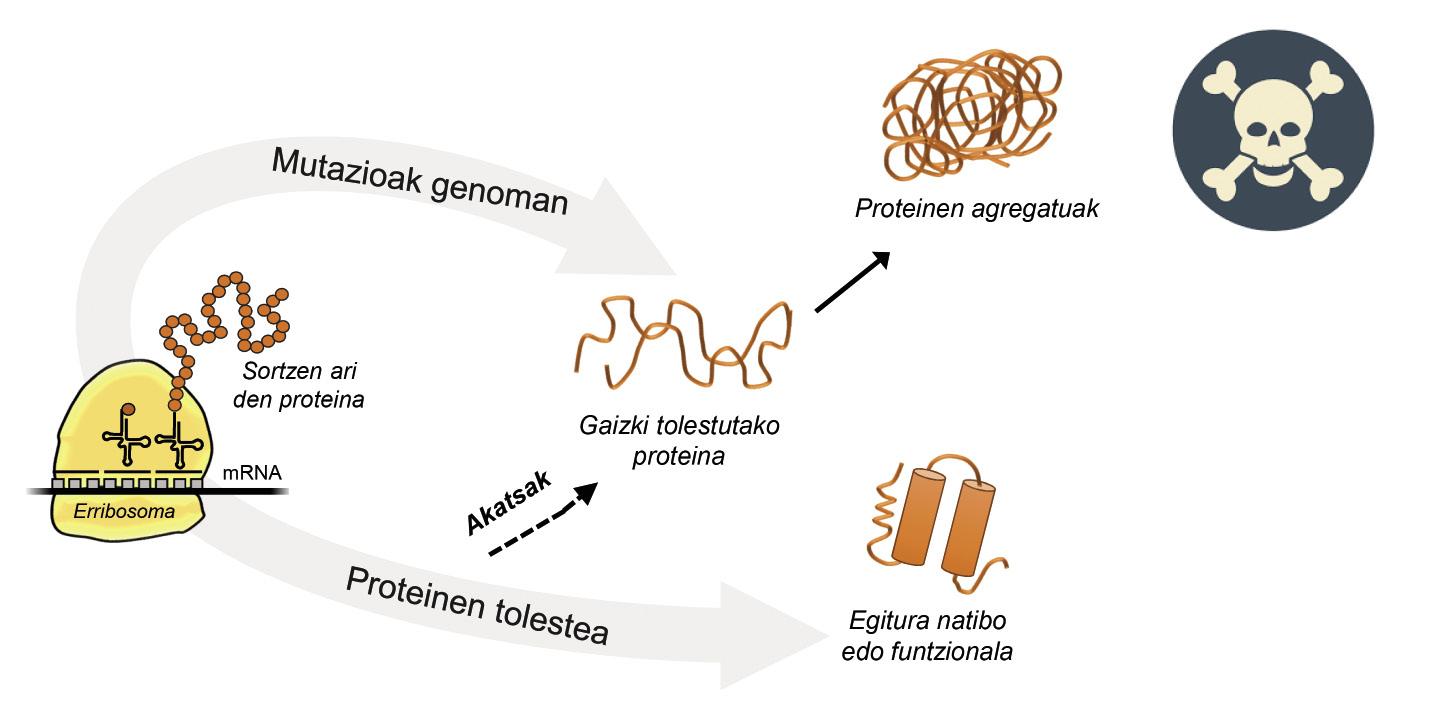
Per exemple, els afectats per la malaltia d'huntington tenen una proteïna d'huntingtina defectuosa. De fet, en mutar-se el gen HTT que codifica la huntingtina, la proteïna produïda conté més aminoàcids glutamina dels necessaris, és a dir, conté massa exemplars d'una perla determinada [4]. Els afectats per l'esclerosi lateral múltiple i la demència frontotemporal també presenten una proteïna C9orf72 defectuosa amb més paris d'aminoàcids glicina dels necessaris, que té una gran tendència a acumular-se en neurones [7].
No obstant això, la malaltia més coneguda per l'acumulació de proteïnes és l'alzheimer, una malaltia que agita fortament la societat actual. A mesura que moren les neurones de l'hipocamp cerebral, les persones malaltes perden les seves capacitats cognitives, especialment la memòria a curt termini. Però, per què moren les neurones? Cada vegada hi ha més evidència que les acumulacions de proteïnes tau i beta-amiloide mutades són les principals responsables de la mort de les neurones [6].
Proteasoma: trituradora de proteïnes
I la cèl·lula no té mecanismes per a degradar proteïnes mal plegades i evitar agregats patològics? Sí, i més d'una. No obstant això, d'una banda, tenint en compte els exemples anteriorment esmentats, és evident que algunes acumulacions de proteïnes són capaces de trencar amb aquests mecanismes, i d'altra banda, la pròpia màquina de degradació de proteïnes pot tenir problemes i impedir el reciclatge.
A més de les proteïnes mal plegades, la majoria de les proteïnes no disponibles per a la cèl·lula es degraden gràcies a un complex multiproteico anomenat proteasoma. Però entre els milers de proteïnes que hi ha en la cèl·lula, com es pot saber quin és el que cal degradar? Com percep el proteasoma quines proteïnes cal reduir? Doncs perquè tenen adherida una petita molècula anomenada ubiquitina. Aquesta molècula funciona com un post-it que diu “has d'anar a proteasoma a degradar”. Proteasoma actua com una trituradora de paper. Redueix les proteïnes, és a dir, trenca les relacions entre els aminoàcids. Paral·lelament, posa a la disposició de la cèl·lula aminoàcids lliures per a la producció de noves molècules [8]. Per això, es pot afirmar que el proteasoma recicla proteïnes (Figura 3).
El procés de reciclatge de proteïnes és molt complex i n'hi ha prou que es produeixi un error en qualsevol punt perquè es desenvolupi una malaltia. En el procés d'unió de la ubiquitina a les proteïnes que seran transferides a proteasoma, col·laboren tres tipus d'enzims: Enzims E1, E2 i E3. Si per estar mutades aquests enzims el procés d'ubiquitinación de les proteïnes no és correcte, les proteïnes a degradar no es destrueixen i s'acumulen en la cèl·lula. Aquesta toxicitat es manifesta sobretot en neurones altament vulnerables, la qual cosa significa que moltes malalties neurològiques estan relacionades amb els defectes que es produeixen en aquests enzims [8].

L'enzim UBA1 tipus E1, per exemple, està relacionada amb una malaltia neuromuscular excepcional que només afecta als nois. Aquests pacients, a més de tenir una gran feblesa muscular, no tenen reflexos, és a dir, els músculs no responen adequadament a estímuls externs. Els que tenen mutada l'enzim UBE2A tipus E2 desenvolupen la síndrome de deficiència UBE2A, una malaltia neurològica atípica. Aquesta malaltia és també patida únicament per homes, sent els símptomes més freqüents la deficiència mental, alteracions cutànies, de l'aparell reproductor i urinari i l'absència de parla.
Entre els tres enzims que realitzen la ubiquitinación de proteïnes, les més abundants són les E3. Així, existeix algun enzim E3 mutada darrere de la majoria de les malalties neurològiques desenvolupades com a conseqüència d'una ubiquitinación defectuosa. Per exemple, els qui sofreixen l'autisme familiar i l'autisme esporàdic, classificats dins dels trastorns de l'espectre de l'autisme, tenen mutades els enzims UBE3B i UBE3C tipus E3, respectivament. Per contra, les persones amb UBE3A defectuosa desenvolupen la síndrome d'Angelman relacionat amb l'autisme [9].
A més de les malalties relacionades amb l'autisme, els E3 mutats poden causar altres malalties neurològiques molt diferents. Per exemple, els qui tenen mutada l'enzim E3 anomenada gigaxonina desenvolupen la neuropatía dels axons gegants. En condicions saludables, la gigaxonina estabilitza els neurofilaments dels braços neuronals, és a dir, dels axons, condicionant la seva estructura i grandària. No obstant això, quan la gigaxonina es muta, es converteix en no funcional i no vascuitina els neurofilaments dels axons. Això fa que no es degradin els neurofilaments i que els axons de les neurones siguin més grans i llargs del necessari. En conseqüència, la majoria dels pacients que sofreixen aquesta neuropatía tenen problemes per a caminar i, en general, per a coordinar els moviments [9].
Després de la recent síndrome de Tenorio es troba un enzim E3, en aquest cas denominada RNF125. Els afectats per la síndrome de Tenorio tenen discapacitat intel·lectual. A més, aquest tipus de pacients es caracteritzen per un creixement excessiu, especialment el desenvolupament excessiu del crani. El contrari els ocorre als que tenen mutat el TRIM36, als quals els falta gran part del crani i del cervell [9].

Conclusions
És evident que un desequilibri en el cicle de síntesi i degradació de proteïnes pot produir efectes realment nocius per a les cèl·lules i els organismes vius en general. D'una banda, proteïnes mal doblegades per mutacions, acumulades com a bosses d'escombraries inutilitzables, poden causar la mort de les cèl·lules. D'altra banda, la deterioració dels dispositius moleculars responsables de la degradació i el reciclatge de proteïnes produeix acumulacions tòxiques de proteïnes. De manera anàloga a l'acumulació d'escombraries en cas que l'activitat d'una instal·lació de reciclatge de residus s'anul·lés.
Sabem que un dels principals reptes de la societat actual és la correcta gestió dels residus. I és que, si no es garanteix el reciclatge dels residus, en pocs anys cobrirem els nostres oceans i camps amb escombraries i la vida es farà insostenible. En el món microscòpic les cèl·lules tenen un repte similar quan tenen problemes per a gestionar proteïnes no funcionals. Per a garantir la seva salut és imprescindible eliminar les proteïnes tòxiques.
Per tant, si la pròxima vegada se t'oblida reciclar els residus de la teva llar, recorda el desastre que això suposaria per a una cèl·lula. De fet, el reciclatge de les escombraries que generem els éssers humans i el reciclatge de les proteïnes que s'acumulen en les cèl·lules són accions similars, a gran i petita escala. Una clau perquè tots puguem mantenir-nos sans.
Bibliografia
Buletina
Bidali zure helbide elektronikoa eta jaso asteroko buletina zure sarrera-ontzian







