



Décoder le génome

Le génome est un code formé par les séquences de quatre nucléotides représentées par les lettres G, A, C et T. Depuis qu'il a été connu, beaucoup de gens ont étudié comment décoder le génome. Et bien sûr, pour décoder, il faut d'abord lire ces lettres ou, autrement dit, séquencer le génome. Autrement dit, il faut savoir dans quel ordre sont ces G, A, C, et ces T.
Pour ce faire, une méthode a surtout été utilisée au cours des 30 dernières années, bien que pendant toutes ces années elle ait connu des améliorations significatives. Cette méthode a été développée par Frederick Sanger et ses compagnons dans les années 1970 -- donc il a reçu le deuxième prix Nobel -.
La réaction de base de la méthode de Sanger exige quatre composants principaux : la partie d'ADN à séquencer ou « modèle d'ADN », quatre types de nucléotides libres, les petits filaments d'ADN de 20-30 nucléotides connus comme initiateurs ou premiers, et l'enzyme d'ADN polymérase qui synthétise l'ADN.
La réaction commence par chauffer l'ADN pour séparer les deux filaments. De cette façon, l'initiateur se joindra à l'un des filaments (où il y a une séquence supplémentaire). Parce que la polymérase DNA allonge les filaments d'ADN, mais on ne peut pas commencer par rien, c'est pourquoi l'initiateur est nécessaire. Une fois que l'initiateur a rejoint le modèle d'ADN, l'ADN polymérase initiateur commence à s'étirer nucléotide : là où il y a un A dans le modèle il mettra un T et vice versa, et là où il y a un G un C et vice versa. Ceci se produit, en outre, toujours dans un sens déterminé, c.-à-d., les deux extrémités d'un filament d'ADN sont appelées 5' et 3', et la polymérase ADN ajoute des nucléotides à l'extrémité 3'.
C'est pourquoi, si nous parlions de plusieurs copies d'un modèle d'ADN, nous obtiendrions autant de copies d'un de ses filaments. Cependant, la clé de la méthode de Sanger est dans l'utilisation de quelques nucléotides modifiés : les didéoxinucléotides. Il manque à ces nucléotides transformés un groupe d'hydroxyles à l'extrémité 3', ce qui fait que d'autres nucléotides ne peuvent pas être ajoutés.

Quatre nucléotides, quatre réactions
La méthode de Sanger nécessite quatre réactions pour chaque modèle d'ADN. Dans les quatre réactions se trouvent les quatre composants mentionnés ci-dessus, mais dans chacun d'eux est placé un dideoxinucléotide unique. Ainsi, par exemple, une des réactions contiendra quatre nucléotides normaux et quelques dioxi-G. Dans cette réaction, lorsque l'ADN polymérase initiateur commence à s'étirer, chaque fois que vous avez besoin d'un G, deux choses aléatoires peuvent se produire: Prendre un G normal ou une dioxi-G. La plupart sont normales G, mais quand vous prenez un dioxi-G, la polymérase ne pourra pas continuer à étirer la chaîne. Ainsi, à partir de plusieurs copies du modèle, nous obtenons des filaments de longueur différente, tous finis dans un G. Et à partir de millions d'exemplaires, nous aurons enfin les filaments correspondant à chaque G de la séquence. Et il en va de même pour les trois autres réactions.
Par la suite, les nouveaux filaments obtenus sont séparés par des tailles par électrophorèse. Dans l'électrophorèse, les filaments d'ADN sont placés dans un champ électrique et comme l'ADN a une charge négative, ils se déplacent du pôle négatif au pôle positif. Sur le chemin se place quelque chose qui rend ce mouvement difficile, comme un gel poreux, de sorte que les grandes parties prennent plus de temps à se déplacer d'un pôle à l'autre que les petites. De cette façon, on peut distinguer les filaments ayant un côté d'un nucléotide.
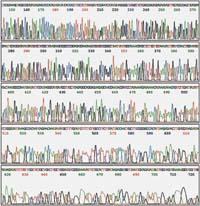
Pour pouvoir visualiser le résultat de l'électrophorèse, les nouveaux filaments doivent être en quelque sorte marqués. Initialement, on utilisait des nucléotides ou initiateurs marqués radialement ou fluorescents. Ainsi, par une autoradiographie, ou par des rayons ultraviolets, ils peuvent être vus comme des bandes, des filaments séparés par des tailles.
Chaque réaction consiste à tracer une ligne dans l'électrophorèse, et le résultat final est que nous verrons une bande par nucléotide de la séquence sur l'une des quatre lignes, et ainsi, suivant l'ordre de ces bandes, nous pourrons lire cette séquence.

Automatisation
C'est essentiellement la technique utilisée pour séquencer la plupart de l'ADN séquencé jusqu'à présent. Mais depuis sa fondation, Sanger a connu d'importantes améliorations. L'un des plus importants était le séquenceur automatique d'ADN inventé par Leroy Hood en 1986. Hood a marqué chacun des quatre dideoxinucléotides afin de fournir une fluorescence de longueur d'onde différente (couleur). Ainsi, d'une part, au lieu de quatre réactions, il suffit d'une seule, et de l'autre, la longueur d'onde émise en étant éclairée par des rayons ultraviolets peut être détectée automatiquement.
Dans les séquenceurs automatiques, chaque échantillon va sur une seule ligne, détectant les quatre nucléotides par leur couleur différente. Dans les séquenceurs modernes, les filaments d'ADN sont séparés en passant par des capillaires de fibre de verre, à la sortie de l'extrémité sont frappés par un laser et la machine détecte la présence de fluoerescence. Les filaments qui sortent d'abord seront l'initiateur plus un nucléotide et ensuite sortira toute la séquence.
Au cours de la dernière décennie, les séquenceurs automatiques d'ADN se sont considérablement améliorés, pouvant traiter beaucoup plus rapidement et faciliter leur utilisation. Actuellement, des centaines d'échantillons peuvent être séquencés en une seule session et en un jour, 24 sessions peuvent être effectuées.
Grand génome

Cependant, il n'est pas encore facile de décoder tout un génome. Les techniques de séquençage de l'ADN servent à séquencer de petits fragments d'ADN, séquences de 300-900 nucléotides. En fait, dans des filaments plus longs, ils ne peuvent pas être séparés avec précision de la face d'un nucléotide.
Mais le petit génome d'une bactérie contient également des millions de nucléotides, et l'humain 3.000.000.000, divisé en 23 chromosomes. Imaginez que pour écrire tout le génome lettre à lettre dans ce magazine nous aurions besoin d'environ 20.000 magazines. La longueur moyenne des séquences lues (500 nucléotides) serait le paragraphe suivant, et sur 20.000 revues on inclut six millions de paragraphes de ce type. En outre, pour obtenir une fiabilité suffisante, une fois non, il faudrait lire entre 6 et 10 fois toutes ces revues.
C'est ce qu'ils ont fait dans le projet de génome humain. Par parties ils ont lu tout le génome humain lettre à lettre. Cependant, diviser tout le génome et lire toutes ces parties est une chose et l'autre est de savoir l'ordre dans lequel ces parties doivent être jointes.
Le projet a commencé par la construction d'une carte de génome. Pour pouvoir parcourir les chromosomes sans perdre, des milliers de points de référence ont été pris. Une fois ces cartes réalisées, une "bibliothèque" de fragments d'ADN a été créée qui engloberait tout le génome. Pour ce faire, ces morceaux d'ADN ont été stockés à l'intérieur des bactéries, et grâce aux repères, chaque partie du génome était connue. En bref, c'est ce que toute bibliothèque offre : informations ordonnées. Cela a permis de travailler de manière coordonnée dans des laboratoires du monde entier.
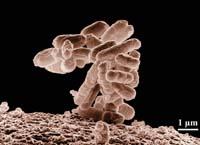
Les fragments d'ADN ont été stockés dans les bactéries E. coli qui vivent habituellement dans nos intestins, comme les chromosomes bactériens artificiels (BAC). Des fragments d'ADN de 100.000 ou 200.000 nucléotides sont stockés dans chaque BAC.
Les bactéries E. coli conservent le temps que vous voulez dans le congélateur. Ainsi, lorsque le scientifique a besoin d'un BAC de la bibliothèque, la bactérie doit seulement ressusciter à 37ºC. En outre, lorsque la bactérie contenant le fragment d'ADN est jouée avec elle. Ainsi, il suffit que les bactéries laissent pousser une nuit pour obtenir des millions de copies de leur partie interne de l'ADN. Ceci est appelé amplification de l'ADN.
Les BACs sont encore très grands pour pouvoir séquencer. Par conséquent, les BACs sont également divisés au hasard, obtenant des parties plus petites qui se chevauchent. Ces parties sont introduites dans les virus ou plasmides qui infectent les bactéries pour une amplification ultérieure dans les bactéries E. coli. Enfin, l'ADN des bactéries est purifié et séquencé. On identifie ensuite les séquences qui se chevauchent dans les différentes parties et on complète la séquence complète du BAC. En même temps, les BACs se chevauchent également. Et ainsi, la lecture par lecture, le BAC par BAC et le chromosome par chromosome, jusqu'à lire tout le génome.
Course génomique
Lire ainsi un génome complet nécessite beaucoup de temps, de travail et d'argent. Par conséquent, les chercheurs étudient continuellement pour développer des techniques de séquençage plus rapides et bon marché. Beaucoup de nouvelles méthodes cherchent à augmenter la capacité de séquençage et réaliser des milliers ou des millions de séquences simultanément. Mais de nouvelles idées ne manquent pas, comme une méthode qui détecte au moment où la dna-polymérase ajoute le nucléotide au filament, ou qui détecte des courants électriques qui identifieraient chaque nucléotide en passant les filaments par nanopors.
Les projets sont nombreux et se développent à une vitesse vertigineuse. Il n'est pas surprenant que les institutions publiques et privées investissent beaucoup. En 2006, le National Human Genome Research Institute (NHGRI) des États-Unis a apporté 13 millions de dollars au financement de projets pour accélérer le développement de technologies couvrant le séquençage de l'ADN. La même année, la Fondation X Prize a annoncé un prix de dix millions de dollars pour la première équipe qui en 2006 réalise un dispositif capable de séquencer cent génomes humains en dix jours. Et, cent non, un projet international qui vient de lancer vise à séquencer les génomes de mille êtres humains.
Il a été dit que le séquençage du génome est l'un des progrès les plus importants dans l'histoire de l'être humain. Des chercheurs du monde entier tentent de dévoiler tous les secrets du génome et de décoder le code. Il est clair que nous sommes à l'ère de la génomique.

Buletina
Bidali zure helbide elektronikoa eta jaso asteroko buletina zure sarrera-ontzian










