Alzheimerren gaixotasuna narriadura kognitiboaren arrazoi nagusia eta gaixotasun neurodegeneratibo ohikoena da (1–3). Gizartearen zahartzearekin zuzen-zuzenean loturik dagoen gaitza da; izan ere, kasuen ia % 95ek 65 urtetik gora izaten dituzte lehen sintomak (4,5). Alois Alzheimer psikiatra alemaniarrak 1906. urtean egin zuen gaixotasun honen lehen deskribapen anatomopatologikoa. Gaur egun, badakigu gaixotasunaren sintomak garatu baina gutxienez 2 hamarkada lehenago amiloide eta tau deritzen proteinen metaketa gertatzen dela burmuinean (6–9).
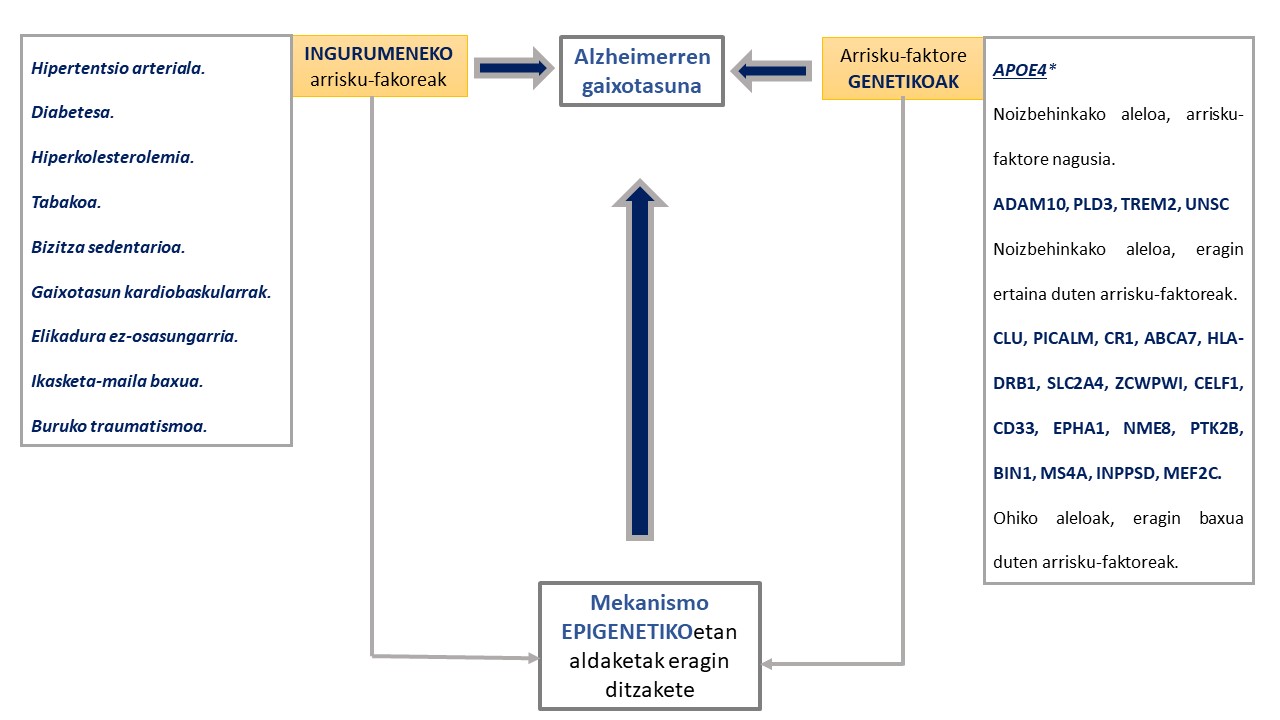
Azken urteotan gaitza garatzeko arriskua areagotzen duten hainbat faktore genetiko eta ingurumeneko deskribatu dira (6,10). Kasu guztien % 1 soilik azaldu daiteke presenilina 1, presenilina 2 eta amiloide proteinaren aitzindariaren geneetan gertatzen diren mutazioen bidez (11,12). Gainerako arrisku faktore genetiko gehienek (apoliproteina E, APOE, -ren salbuespena medio) eragin txikia edota eztabaidagarria dute gaixotasunaren garapenean (4,6,8,13)(1.Irudia).
Epigenetika
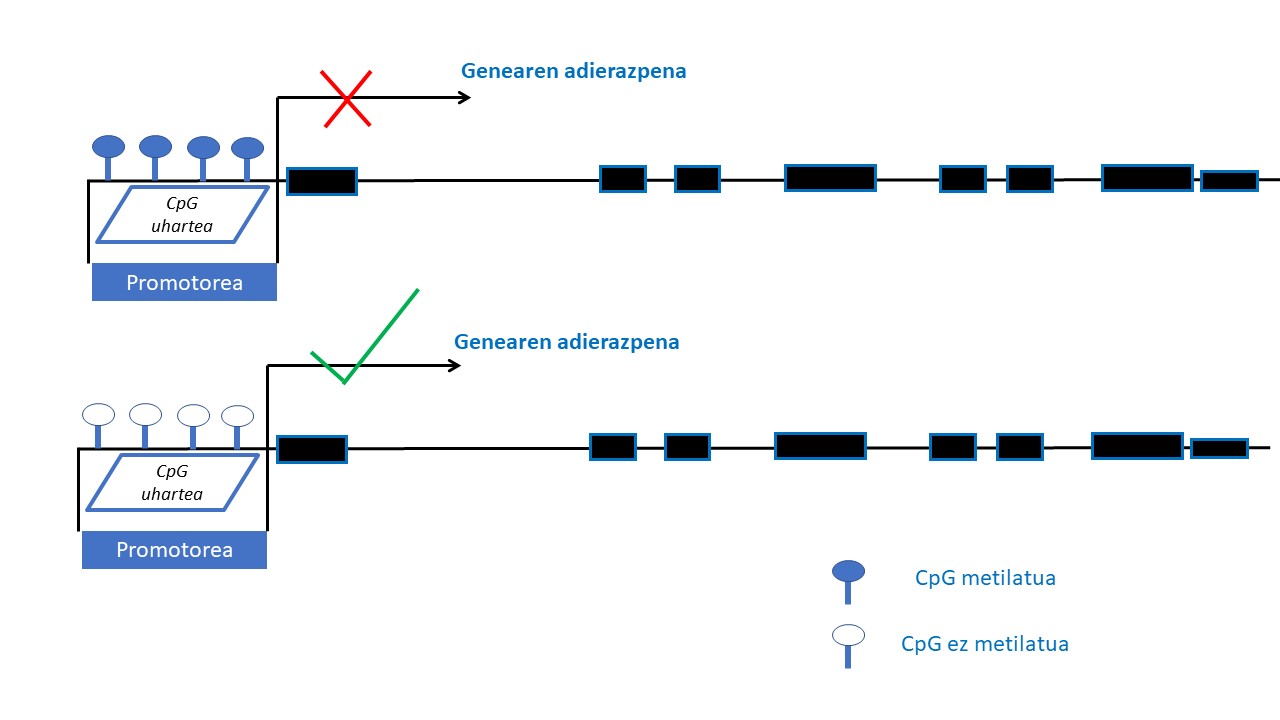
Epigenetika geneen adierazpena azido desoxirribonukleikoaren (DNA) sekuentzia aldatu gabe moldatu dezaketen mekanismoak ikertzeaz arduratzen da. Mekanismo epigenetiko horiek heredagarriak dira, baina, era berean, ingurumeneko zenbait faktorek aldaketak eragin ditzakete (14). Mekanismo epigenetiko ikertuena DNAren metilazioarena da. DNAren metilazio-prozesuan, metilo-talde bat genearen 5’ kokapenan dagoen zitosina bati elkartzen zaio, metilzitosina sortuz. Metilazioa guaninaz jarraiturik dauden zitosinetan gertatzen da batez ere, CpG deritzan diklunetidoetan zehazki. CpG dinukleotido horiek bereziki geneen adierazpena erregulatzeaz arduratzen diren eremuetan dira ohikoen, promotor-eremuetan esaterako, eta CpG uharteak osatzen dituzte han (eremu batean, DNAren sekuentziaren % 50 baino gehiago CpGak direnean). Genearen promotore-eremuan CpG uharteak metilatuak egoteak gene horren adierazpena isiltzen du eta aldiz, CpG uharteak eremu berean metilaturik gabe badaude genearen adierazpena posible izango da (15) (2. Irudia).
DNAren metilazioaren eragina Alzheimerren gaixotasunean
Alzheimerren gaixotasuna oso gaitz konplexua da, eta kasu gehienetan hainbat arrisku-faktore genetiko eta ingurumenekoen interakzioaren ondorio da. DNAren metilazioaren ikerketak lagundu diezaguke gaixotasunaren oinarria eta arrisku-faktore genetiko eta ingurumenekoen interakzioaz gehiago ikasten. Zentzuzkoa da Alzheimerren gaixotasunaren behin-betiko diagnostiko anatomopatologikoa duten gaixoen eta anatomopatologikoki gaixotasun neurodegeneratiboen zantzurik ez dituzten burmuinetako hipokanpoak ikertzea, ikusteko ea aldaketak dauden DNAren metilazioan. Lan ugari (16) argitaratu dira azken urteotan aztertzen dutenak zein den DNAren metalizazioaren patroia burmuineko zenbait eremutan, baina ez hipokanpoan. Memoriaren eginkizunerako ezinbesteko egitura da hipokanpoa, bai eta tau proteinaren metaketa hasten den eremua ere (17). Tau proteinaren metaketa gehiago lotzen da sintomen garapenarekin amiloidearen metaketa baino (18).
Gaur egun, zuzenean ikertu daiteke gene jakin bat erregulatzeko eginkizuna duen eremuaren metilazioa (gene kandidatoaren estrategia). Era berean, osoki aztertu litezke burmuineko eremu zehatz batean genoma osoan (genome wide deritzo estrategia horri) dauden metilazio-kantitatearen ezberdintasunak (metilazioaren karakterizazioa). Bi estrategiok osagarriak dira, eta bakoitzak baditu bere abantailak.
- Hipokanpoaren metiloma Alzheimerren gaixotasunean
Nafarroako burmuin-bankuko 38 giza hipokanpo baliatuz (26 Alzheimerren gaixotasuna zutenenak eta 12 gaixotasun neurodegeneratiborik gabekoenenak), Alzheimerren gaixotasunaren hipokanpoko metiloma ikertu dugu lehen aldiz. Illumina enpresaren kit komertziala erabiliz eta kalitatea-kontrola pasatu ostean, 264.031 CpG-tako metilazio-mailak aztertu ditugu. Metilazio-maila ezberdineko 118 CpG posizio detektatu ditugu, 159 geneetatik gertu, CpG irletan kokatuak gehienak. Horien % 86k metilazio-maila altuagoak dituzte Alzheimerren gaixotasuna dutenen artean. Emaitzak bisulfito-sekuntziazioaren bitartez baliozkotu ditugu.
Alzheimerren gaixotasunean metilazio-maila altuagoak dituzten posizioen % 66,6k bat egiten dute kromatina bibalentea daukaten geneen erregulazio-eremuekin. Kromatina bibalentea duten erregulazio-eremu horiek isildurik egoten dira gehienetan, baina, beharrezko izanez gero, edonoiz aktibatzeko prest daude, batik bat erregulazio-funtzioak dituzten geneetan daude, eta garrantzi handia dute zelulen bereizketa-prozesuan. Era berean, sistema semiautomatiko kuantitatibo baten bitartez (Image J softwarea), ikusi dugu metilazio-maila ezberdinak dituzten posizioak hipokanpoko tau proteinaren kantitatearekin erlazionaturik daudela. Gainera, lehenik Pubmed bilatzailean eskuz eta, gero, tresna bioinformatikoen bitartez (GREAT tresna), ikusi dugu metilazio-maila ezberdineko posizioei loturik dauden geneen % 42,4 ezinbestekoak direla nerbio-sistemaren garapenean edo neurogenesian. Homeobox deritzan transkripzio-faktoreak ere hipermetilatuak daude Alzheimerren gaixotasuna pairatzen duten giza hipokanpoetan. Azken horiek garrantzi handia dute embriogenesi-prozesuan, bai eta zelulen desberdintze pluripotentzialean eta, nola ez, neurogenesian ere (19)(3. irudia).
.jpg)
Interesgarria da ikustea Alzheimerren gaixotasunaren baitan metilazio-maila ezberdinak neurogenesiari loturik daudela, eta ez, ordea, neurodegenerazioari (ez apoptosiari, ez autofagiari, ez estres oxidatiboari ezta mitokondrien funtzioari ere). Izan ere, giza hipokanpo osasuntsuan neurogenesia bizitza osoan gertatzen da, eta kalkulatzen da hipokanpoko neuronen herena etengabe berritzen dela. Hipokanpoko neurogenesia gutxitu egiten da adinarekin, eta badirudi are gehiago gutxitzen dela Alzheimerren gaixotasuna bezalako gaitz neurodegeneratiboen baitan (20–23). Lan honen hipotesia da mekanismo epigenetikoek, zehazki metilazioak, Alzheimerren gaixotasunarean neurogenesiaren disregulazioan eragin dezakeela. Lan hori argitaratu zen arte, Homeobox transkripzio-faktoreak ez ziren aurrez Alzheimerren gaixotasunarekin lotu.
- PLD3 genearen metilazio-maila Alzheimerren gaixotasuna duten hipokanpoetan
Gene hautagaiaren estrategia erabiliz, Alzheimerren gaixotasuna garatzeko arriskua ustez areagotu dezakeen gene bat ikertu dugu. Gene horren polimorfismo zehatz bat (gutxienez gizartearen % 1ek daukan DNA-sekuentziaren aldaera bat) gaitza garatzeko arrisku-faktore gisa deskribatuta zegoen (24), nahiz eta asoziazio hori eztabaidagarritzat jotzen zen (25–28). PLD3, fosfolipasa 3 deritzana, giza burmuinean adierazten den gene bat da, hipokankoan, bereziki. Badiru amiloide proteinaren aitzindariaren prozesaketan garrantzia izan dezakeela (25).
Nafarroako burmuin-bankuko 54 giza hipokanpo erabili ditugu: 36 Alzheimerren gaixotasuna estadio ezberdinetan zuten gaixoenak, eta 18 gaixotasun neurodegeneratibo diagnostiko gabekoenak. PLD3aren azido erribonukleiko mezulariaren (RNAm) mailak neurtu ditugu polimerasaren kate-erreakzioaren metodo kuantitatibo baten bitartez (RT-PCR).
Alzheimerren gaixotasuna zuten hipokanpoetan, PLD3-aren RNAm-a gutxiago adierazten da, eta ezberdintasunak are nabarmenagoak dira gaixotasunaren azken estadioetan. Gainera, ikusi dugu alderantzizko erlazio esanguratsua dagoela PLD3aren mRNAren eta hipokanpoan pilaturik dagoen tau proteinaren kantitatearen artean. Ondoren, PLD3 genearen 2 eremu promotoreetan metilazio-mailak ikertu ditugu, bai pirosekuentziazioen eta bai bisulfitoen sekuentziaren bidez. Ez dugu ezberdintasunik aurkitu promotore nagusian, baina bai, ordea, promotore alternatiboan. Azken horretan, metilazio-maila altuagoa da Alzheimerren gaixotasuna duten hipokanpoetan (29)(4. irudia).
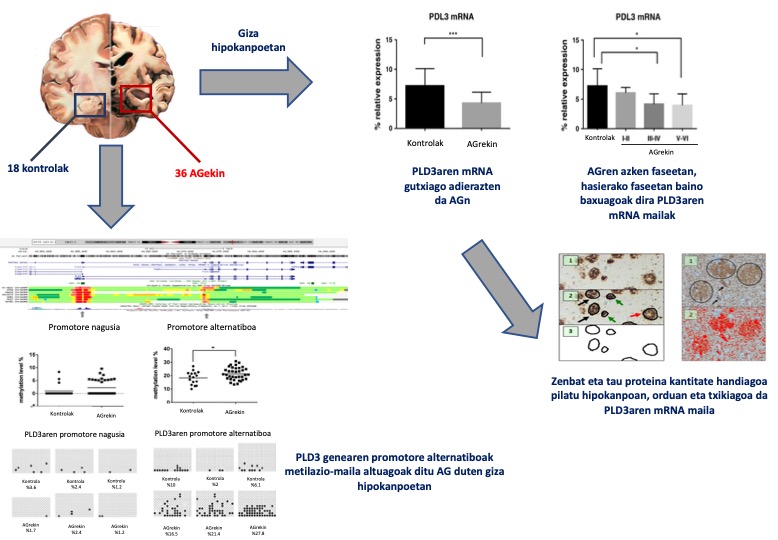
Lan honen bidez, PLD3 geneak Alzheimerren gaixotasunean izan dezakeen garrantzia beste ikuspuntu batetik ikertu dugu. Ez dugu aurrez arrisku-faktore gisa identifikatutako polimorfismoaren presentzia neurtu. Gure kasuan, gene honen adierazpena erregulatzen duen metilazio-mailan ezberdintasunak ikusi ditugu, eta, gainera, erlazioa topatu dugu tau proteinaren mailekin. Gure aurkikuntzek berresten dute PLD3 genearen eginkizun patogenikoa, polimorfismo zehatz batetik haratago. Era berean, metilazio-mailan ezberdintasunak promotore alternatiboan egoteak —eta ez nagusian—mekanismo epigenetikoen erregulazioaren konplexutasunaren berri ematen digu.
Ondorioak
Epigenetikak Alzheimerren gaixotasunaren patogenia ulertzen lagundu diezaguke. Alde batetik, erraztu dezake aurrez arrisku-faktore genetiko eztabaidagarritzat hartutakoen eginkizun patogenikoa ikertzea. Bestetik, bide ematen du pentsatzeko Alzheimerren gaixotasunaren baitan hipokanpoan gertatzen den neurogenesiaren galera, neurri batean behintzat, metiloman gertatzen diren aldaketen ondorio izan litekeela.
Ingurumen-faktoreek, horien artean botikek, metilazioan gainerako mekanismo epigenetikoetan bezala eragin dezakete, eta hori gure mesederako erabil genezake. Beraz, epigenetikak, Alzheimerren gaixotasunaren oinarria hobeto ulertzeaz gain, jomuga terapeutiko berriak garatzen lagundu diezaguke.
Hala ere, kontuan izan behar dugu epigenetikaren baitan asko daukagula ikasteko oraindik, eta, eguneroko klinika praktikoan aplikatzen hasi aurretik, ezinbestekoa izango dela burmuineko eremu ezberdinetan —eta idealki likido zefalorrakideoan edota odolean— atzeman dezakegun sinadura epigenetiko komun bat ezagutzea.
Bibliografia
1. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CRJ, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011 May;7(3):263–9.
2. Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology. 2013 May;80(19):1778–83.
3. Zetterberg H. Applying fluid biomarkers to Alzheimer’s disease. Am J Physiol Cell Physiol. 2017 Jul;313(1):C3–10.
4. Rabinovici GD. Late-onset Alzheimer Disease. Continuum (Minneap Minn). 2019 Feb;25(1):14–33.
5. Castro DM, Dillon C, Machnicki G, Allegri RF. The economic cost of Alzheimer’s disease: Family or public health burden? Dement Neuropsychol. 2010;4(4):262–7.
6. Dubois B, Hampel H, Feldman HH, Scheltens P, Aisen P, Andrieu S, et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016 Mar;12(3):292–323.
7. Niemantsverdriet E, Valckx S, Bjerke M, Engelborghs S. Alzheimer’s disease CSF biomarkers: clinical indications and rational use. Acta Neurol Belg. 2017 Sep;117(3):591–602.
8. Lane CA, Hardy J, Schott JM. Alzheimer’s disease. Eur J Neurol. 2018 Jan;25(1):59–70.
9. Blennow K, Zetterberg H. Biomarkers for Alzheimer’s disease: current status and prospects for the future. J Intern Med. 2018 Dec;284(6):643–63.
10. Molinuevo JL, Ayton S, Batrla R, Bednar MM, Bittner T, Cummings J, et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 2018 Dec;136(6):821–53.
11. Schindler SE, Fagan AM. Autosomal Dominant Alzheimer Disease: A Unique Resource to Study CSF Biomarker Changes in Preclinical AD [Internet]. Vol. 6, Frontiers in Neurology . 2015. p. 142. Available from: https://www.frontiersin.org/article/10.3389/fneur.2015.00142
12. Bateman RJ, Aisen PS, De Strooper B, Fox NC, Lemere CA, Ringman JM, et al. Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res Ther. 2011 Jan;3(1):1.
13. Fernández-Viadero C, Rodríguez Rodríguez E, Combarros Pascual O, Crespo Santiago D. [Genetics and Alzheimer’s disease: a population at risk]. Rev Esp Geriatr Gerontol. 2013;48(1):39–44.
14. Oh G, Ebrahimi S, Wang S-C, Cortese R, Kaminsky ZA, Gottesman II, et al. Epigenetic assimilation in the aging human brain. Genome Biol. 2016 Apr;17:76.
15. Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008 Jun;9(6):465–76.
16. Mendioroz Iriarte M, Pulido Fontes L, Méndez-López I. [Neuroepigenetics: Desoxyribonucleic acid methylation in Alzheimer’s disease and other dementias]. Med Clin (Barc). 2015 May;144(10):457–64.
17. Jaroudi W, Garami J, Garrido S, Hornberger M, Keri S, Moustafa AA. Factors underlying cognitive decline in old age and Alzheimer’s disease: the role of the hippocampus. Rev Neurosci. 2017 Oct;28(7):705–14.
18. Arnsten AFT, Datta D, Del Tredici K, Braak H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimers Dement. 2021 Jan;17(1):115–24.
19. Altuna M, Urdánoz-Casado A, Sánchez-Ruiz De Gordoa J, Zelaya MV, Labarga A, Lepesant JMJ, et al. DNA methylation signature of human hippocampus in Alzheimer’s disease is linked to neurogenesis. Clin Epigenetics. 2019;11(1).
20. Moreno-Jiménez EP, Flor-García M, Terreros-Roncal J, Rábano A, Cafini F, Pallas-Bazarra N, et al. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat Med. 2019 Apr;25(4):554–60.
21. Gilbert G. Neurogenesis takes a hit in Alzheimer’s disease. Sci Transl Med [Internet]. 2019 May 1;11(490):eaax1726. Available from: https://doi.org/10.1126/scitranslmed.aax1726
22. Tobin MK, Musaraca K, Disouky A, Shetti A, Bheri A, Honer WG, et al. Human Hippocampal Neurogenesis Persists in Aged Adults and Alzheimer’s Disease Patients. Cell Stem Cell. 2019 Jun;24(6):974-982.e3.
23. Scopa C, Marrocco F, Latina V, Ruggeri F, Corvaglia V, La Regina F, et al. Impaired adult neurogenesis is an early event in Alzheimer’s disease neurodegeneration, mediated by intracellular Aβ oligomers. Cell Death Differ. 2020 Mar;27(3):934–48.
24. Cruchaga C, Karch CM, Jin SC, Benitez BA, Cai Y, Guerreiro R, et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature. 2014 Jan;505(7484):550–4.
25. Satoh J-I, Kino Y, Yamamoto Y, Kawana N, Ishida T, Saito Y, et al. PLD3 is accumulated on neuritic plaques in Alzheimer’s disease brains. Alzheimers Res Ther. 2014;6(9):70.
26. Cacace R, Van den Bossche T, Engelborghs S, Geerts N, Laureys A, Dillen L, et al. Rare Variants in PLD3 Do Not Affect Risk for Early-Onset Alzheimer Disease in a European Consortium Cohort. Hum Mutat. 2015 Dec;36(12):1226–35.
27. Lambert J-C, Grenier-Boley B, Bellenguez C, Pasquier F, Campion D, Dartigues J-F, et al. PLD3 and sporadic Alzheimer’s disease risk. Vol. 520, Nature. England; 2015. p. E1.
28. Heilmann S, Drichel D, Clarimon J, Fernández V, Lacour A, Wagner H, et al. PLD3 in non-familial Alzheimer’s disease. Vol. 520, Nature. England; 2015. p. E3-5.
29. Blanco-Luquin I, Altuna M, Sánchez-Ruiz de Gordoa J, Urdánoz-Casado A, Roldán M, Cámara M, et al. PLD3 epigenetic changes in the hippocampus of Alzheimer’s disease. Clin Epigenetics. 2018 Sep;10(1):116.
Miren Altuna Azkargorta
CITA-Alzheimer Fundazioa (Donostia) eta Neuroepigenetikako laborategia, Navarrabiomed (Iruñea)